- About us
- Research
- Students & Teaching
- Seminars & Events
- Directories
- Booking Rooms & Equipment
- עברית
Home » Prof. Batsheva Kerem has developed a new drug for the treatment of cystic fibrosis patients
Cystic fibrosis (CF) is a life-shortening multi-organ genetic disease affecting ~ 90,000 individuals worldwide, caused by mutations in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene that encodes a chloride (Cl-) channel located at the surface of epithelial cells. The impaired CFTR activity alters electrolytes and hydration balance across epithelia, causing the accumulation of thick mucus in the lung bronchial tree leading to a chronic progressive lung disease, which is the major cause of morbidity and mortality.
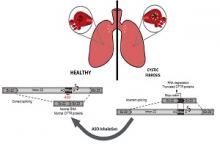
In the present study, together with SpliSense Therapeutic we aimed to develop a new therapy for CF patients carrying the 3849+10kb C-to-T splicing mutation, leading to aberrant processing of the RNA, termed splicing. The mutation leads to aberrant inclusion of a specific sequence into the RNA molecules which leads to degradation of the RNA, as well as to the production of truncated non-functional CFTR proteins (see figure). The 3849+10kb C-to-T defect is the 7th most common CFTR defect in the US and 8th in Europe, carried by ~1600 CF patients. In several populations, the mutation is highly prevalent, such as in Ashkenazi Jews and CF patients in Slovenia, Poland and Italy. The current available CFTR modulator drugs have only a limited clinical benefit for patients carrying splicing mutations including the 3849+10kb C-to-T mutation. Thus, further strategies of drug development are essential to address the unmet needs of patients carrying this mutation.
We aimed to identify molecules termed antisense oligonucleotides, that are complementary to specific sequences along the CFTR gene carrying this mutation, which can correct the RNA defective processing. SpliSense has screened ~30 antisense molecules in a cell model. The effect of highly potent candidate molecules on the CFTR RNA processing and protein function was further analysed in human respiratory cells, derived from various patients carrying at least one copy of the 3849+10kb C-to-T mutation. We were able to efficiently deliver the antisense oligonucleotides into the cells by free uptake, without any carriers. In addition, we have used molecules with different chemical structures to identify the most efficient and potent molecule.
Following comprehensive analyses, in various cellular systems, we have identified a lead drug candidate, able to correct the aberrant CFTR splicing and restore normal protein function in respiratory cells from patients carrying the 3849+10kb C-to-T defect. Optimized efficiency was further obtained using different chemical structures of the molecule. The lead drug candidate was highly effective and potent and was able to restore the CFTR channel function to levels expected to confer a significant clinical benefit, and improved quality of life for patients carrying this defect.
The presented results lead to SpliSense current clinical development of inhaled therapy of the lead molecule.