Just like people, proteins interact with other proteins and participate in “social networks”. Some proteins are more social, having a greater number of interaction partners while other proteins prefer to have only a few good friends.
Each protein-protein interaction is characterized with certain binding affinity or binding free energy. Mutations in proteins can change this affinity, sometimes abrogating and sometimes stabilizing the interaction. The change in binding affinity of one protein-protein interaction could translate into remodeling of the whole protein network, frequently leading to dysregulation of the network and disease.
Therefore, understanding how specific mutations in protein complexes affect their binding affinity is extremely important to both basic biology and to biomedical sciences, where inhibition of a particular protein-protein interaction might help to treat the related disease.
The existing approaches for measuring changes in binding affinity due to mutations are labor-intensive: it could take months to years to obtain such data for hundreds of protein mutants.
Michael Heyne from the Shifman lab in collaboration with Niv Papo at the Ben Gurion University developed a novel high-throughput approach that allows us to obtain the quantitative binding affinity data for thousands of protein mutants in one experiment. Our approach combines yeast surface display technology, deep sequencing and data normalization and produces binding affinity data that is in high agreement with low-throughput experiments.
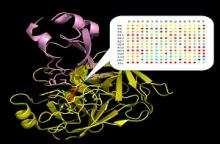
Using this powerful methodology, we measured binding affinity for all single mutants one exemplary complex between bovine trypsin and its inhibitor BPTI. We demonstrate that this protein-protein complex is highly optimized evolutionary with only one mutation leading to affinity improvement and many single mutations completely destroying the interaction.
The application of our approach to multiple protein complexes and comparison of how mutations affect their binding affinities would help us to understand how protein evolve. In addition, our approach could be used in various drug design efforts, where protein-based drugs are engineered and affinity-matured for interaction with their targets.